Science
The Spencer lab studies the organization and function of the human genome and how altered genome function contributes to acute myeloid leukemia.
 Our group specializes in cancer genetics, genomics, and
epigenetics with a focus on AML. We are specifically focused on acute myeloid leukemia
(AML), a devastating cancer that affects children and adults. Despite major
advances in the molecular understanding of this disease, current
therapies have changed little over the past 30
years. We aim to understand how mutations and epigenetic changes alter the structure
and function of the genome in ways that contribute to AML development, progression,
and relapse.
Our group specializes in cancer genetics, genomics, and
epigenetics with a focus on AML. We are specifically focused on acute myeloid leukemia
(AML), a devastating cancer that affects children and adults. Despite major
advances in the molecular understanding of this disease, current
therapies have changed little over the past 30
years. We aim to understand how mutations and epigenetic changes alter the structure
and function of the genome in ways that contribute to AML development, progression,
and relapse.
Current research projects
HOX gene regulation in AML
 HOX genes are conserved
transcription factors first discovered in Drosophila that regulate normal
self-renewal in mammalian hematopoietic cells and are highly expressed
in human AML cells. Transcriptomic profiling of nearly 200 primary AML samples
by our group showed that HOXA and/or HOXB genes are highly expressed
in more than half of all AML patients and are therefore among the most
common drivers of self-renewal in AML cells (Spencer et al., Leukemia
2015). While progress has been made in understanding—and
targeting—HOX gene regulation in some rare AML types, effective
therapeutic strategies for most patients have not been
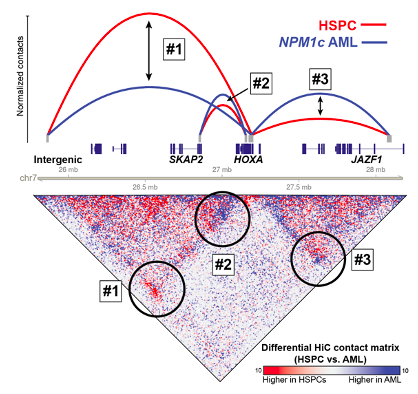
identified. We have investigated the 3D genome architecture of the HOXA cluster in
AML cells with mutations in NPM1 and whether specific CTCF binding sites are
critical for maintaining HOXA genomic interactions and gene
regulation. We found that these CTCF sites are not absolutely required for
HOXA gene expression, which appears to be mainted by specific
interactions with putative HOXA enhancers
(Ghasemi et. al., Leukemia 2020). We
subsequently showed that certain interactions that were maintained
when CTCF sites were deleted displayed AML-specific patterns of DNA methylation
and histone modifications and therefore may represent AML-specific
HOXA enhancers. We are now using CRISPRi in AML samples and massively
parallel reporter assays in cell lines and primary AML samples to
define the cis-acting regulatory sequences that are critical for HOXA
expression in AML.
HOX genes are conserved
transcription factors first discovered in Drosophila that regulate normal
self-renewal in mammalian hematopoietic cells and are highly expressed
in human AML cells. Transcriptomic profiling of nearly 200 primary AML samples
by our group showed that HOXA and/or HOXB genes are highly expressed
in more than half of all AML patients and are therefore among the most
common drivers of self-renewal in AML cells (Spencer et al., Leukemia
2015). While progress has been made in understanding—and
targeting—HOX gene regulation in some rare AML types, effective
therapeutic strategies for most patients have not been
identified. We have investigated the 3D genome architecture of the HOXA cluster in
AML cells with mutations in NPM1 and whether specific CTCF binding sites are
critical for maintaining HOXA genomic interactions and gene
regulation. We found that these CTCF sites are not absolutely required for
HOXA gene expression, which appears to be mainted by specific
interactions with putative HOXA enhancers
(Ghasemi et. al., Leukemia 2020). We
subsequently showed that certain interactions that were maintained
when CTCF sites were deleted displayed AML-specific patterns of DNA methylation
and histone modifications and therefore may represent AML-specific
HOXA enhancers. We are now using CRISPRi in AML samples and massively
parallel reporter assays in cell lines and primary AML samples to
define the cis-acting regulatory sequences that are critical for HOXA
expression in AML.
DNA methylation and 3D genome architecture in AML cells
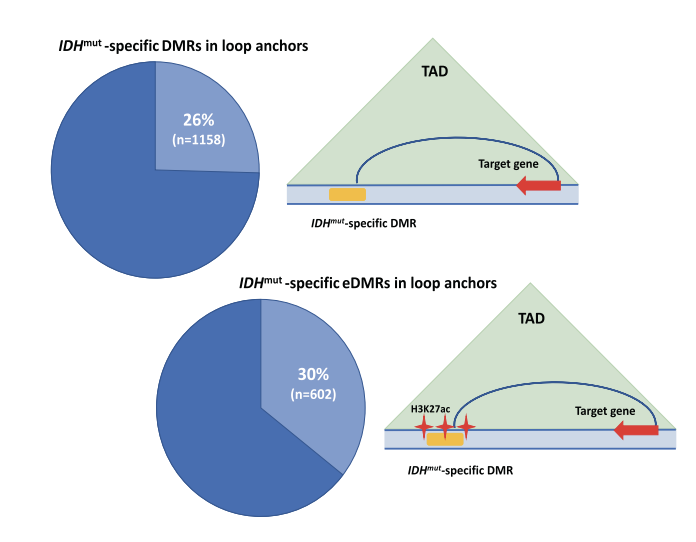
DNA methylation is the most commonly mutated pathway in AML. DNMT3A mutations result in hypomethylation and mutations in IDH1, IDH2, and TET2. We have chacterized the DNA methylation phenotype of primary AML samples with mutations in IDH1 and IDH2 that identified ~4,000 regions with discrete focal hypermethylation that are highly enriched for H3K27 acetylation ChIP-seq peaks and loop anchors in Hi-C data from AML samples indicating that regulatory enhancers are targets for altered methylation in IDH mutant AML cells. We are currently using CRISPR/Cas9 to edit the genome and epigenome of AML cells to understand how altered DNA methylation affects enhancers and gene expression in AML.